1. Explanation
General
NON-SMOKERWEB is a Hauser-Feshbach statistical model code optimized for an energy and temperature range which is interesting for astrophysical purposes. It is related to the well-known SMOKER and NON-SMOKER codes.Calculations in the statistical model require the input of a number of different nuclear properties:
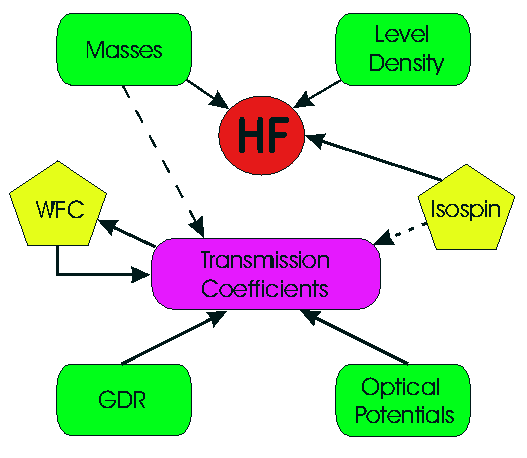
Although the model is well-established, different calculations vary in the various inputs and treatments of such properties. In the calculations presented here, it was attempted to find descriptions which can be used to predict nuclear properties also for nuclei far off stability for which no experimental data is known. Therefore, it was tried to minimize the direct input of experimental data (although some parametrizations are fit to available nuclear data). Thus, we are interested in descriptions which lead to a low average deviation between predicted and observed properties. Nevertheless, local deviations of higher magnitude can occur.
Different options can be chosen as input to the statistical model calculation. The default values are those used in the calculations published in ADNDT 75 (2000) 1 and 79 (2001) 47 (except for experimental masses which have been updated since). For a more detailed explanation of the options see those papers and the reference section.
Important note: The default settings of the code make use of global inputs, attempting to include as few local information as possible. For a fair comparison to other codes be aware that many other codes include a lot of local information (such as level densities or GDR properties) for nuclei at stability. When performing such comparisons, the NON-SMOKERWEB settings should be chosen accordingly!
Registration
To run NON-SMOKERweb the user has to be registered. If you want to start a collaboration and use NON-SMOKERweb please use the Registration Form, specifying in detail what you want to use the code for. You will then be sent a username and password shortly. The access may be restricted to certain targets or projectiles and certain options.Running NON-SMOKERweb
The HTML interface should be quite self-explanatory. In the element box, the standard abbreviation of the element should be input, as it is used in periodic systems or nuclear charts; e.g.Ne for Neon or au
for Gold (input is not case
sensitive). The massnumber is an integer with 2 or 3 digits.
Options and Output
The recommended set of options is the default, therefore it should be sufficient in most cases to just enter element and mass number and to select which kind of projectile one wants to use. The output will contain the nuclear reaction cross sections for three different channels (gamma, n, p, alpha) depending on which projectile was chosen. It will also contain the astrophysical reaction rates. For reactions with a neutral projectile, the Maxwellian averaged cross sections (MACS) are additionally written whereas for charged particle induced reactions the astrophysical S-factor is additionally written. For astrophysical applications it is not only necessary to know the reactions on targets in the ground state but also on thermally excited targets. The reaction rates and MACS are both given for g.s. and thermally excited targets, cross sections and S-factors only for targets being in the g.s.Theoretical masses:
By default, in the calculations experimental values are used if
available (for the current status of the experimental masses included,
see also the history section.
The setting of the theoretical mass will affect
experimentally unknown masses (separation energies), as well as the
level density for all nuclei if the default level
density is
chosen.
The FRDM covers the widest mass range, ETFSI values are available only
for A>35, and the ETFSIQ table starts at Cr (Z>23).
Hint: The output can be saved by right-clicking into the returned page and then selecting "Save as..." and the appropriate format (usually, that is text format). This yields a nice text file which can be inspected further. (For further hints, see below.)
Custom input file
Using a custom input file offers even more flexibility than the options directly provided on the web page. In order to use a file, the appropriate option on the web page has to be checked and a file location has to be provided in the appropriate field. On submission of the web form, the file will be loaded and the appropriate data used (if the file contains more than the data required by the checked options, the excess data will be ignored). For each calculation, the input file will be loaded again from the specified location. The input file is an ASCII file which contains options and data in a specific format. Invalid input files will trigger error messages.Input file format:
- Basics:
- The input file is a plain ASCII file (text file). The name of the file is irrelevant but some browsers do not assume to be sending a plain text file when the extension is something else than .txt, .dat, or .inp and they might encode it inappropriately. Therefore one of these file extensions should be used preferentially. Take a look at the sample input file (the data contained therein is just for illustrative purposes and does not have any real physical meaning) and modify it for your own purposes.
- The maximum length of a line in the input file is 500 characters. If that length is exceeded, errors or unwanted behavior of the code may result!
- Data is organized into sections which start with an identifier and end with the general end-identifier END. The order of the sections is irrelevant but they have to be between the INPUT_START and INPUT_END labels. Everything outside of these labels or between sections is ignored.
- NON-SMOKERweb uses input only from sections which correspond to the options selected on the web form. All other data is ignored.
- Section labels (identifiers) and special labels start with '#*'. They are case sensitive!
- Comment lines use an '#' as first character (and don't have an '*' as second character) and can appear everywhere. Blank lines are ignored.
- Some details:
- Optical potentials:
- Section labels: CUSTOM_ALPHA, CUSTOM_NEUTRON, CUSTOM_PROTON.
- For the proton and alpha potentials, the first
entry in the section
defines the Coulomb radius rc.
The value of the
radius has to follow the label on the same line:
#*RCOUL 1.2
(For the neutron potential there is no such line; it is an error if such a line is present.) - Within these sections there are two subsections each (both have to be specified): The real part of the optical potential is defined from the beginning of the section to the label REALEND; the imaginary part is defined after the label REALEND up to the END identifier.
- Within each of the two subsections, there are two ways to specify the potential: Either by explicitely giving data points or by providing an algebraic expression to compute the potential. The first method is selected with the label DATA, the second with EXPRESSION. Each of these labels have to be given before any data or expression in that subsection.
- Option DATA: Pairs of radius (in fm) and potential depth (in MeV) are given. At most 800 pairs can be given. They don't have to be equidistant but have to be in ascending order by radius (and all radii have to be different). Points between the given data are linearly interpolated. At least two pairs have to be given explicitly. Above and below the first and last point, respectively, the potential is extrapolated using the same slope as between the last two known points. (If you want the potential to become constant, specify a depth twice.)
- Option EXPRESSION: Specify a function to compute the optical potential part at a given radius. See the detailed explanation of custom potentials below. Any whitespace characters (including blanks, tabs, newlines) are ignored, they can be used to structure the input for better (user) legibility. The current upper length limit is 500 characters (not counting whitespace).
- Level density:
- Section label: CUSTOM_LEVEL_DENSITY
- Custom parameters for a shifted Fermi-gas description of the nuclear level density can be provided here. The order of parameters is charge Z, mass number A, level density parameter a (in MeV-1), shift Delta (in MeV).
- During a calculation the default level density (see web page) is used unless Fermi-gas paramaters have been specified for that nucleus. In the latter case a Fermi-gas level density with those parameters is used. At low excitation energies, a constant temperature formula is used in both cases to avoid the divergence of the Fermi-gas description.
- Masses
- Section label: MASSES
- Each entry contains charge Z, mass number A, and the mass excess (in MeV).
- During a calculation the default mass (either experimental or theoretical, see web page) is used unless the mass excess for that nucleus has been specified in the input file. An explicitly defined mass excess is always used when it is found in the input file.
- To change a separation energy, specify the proper two masses but keep in mind that an altered mass also may affect the separation energies in other reaction channels.
- Excited states:
- Section label: EXCITED_STATES
- Low-lying excited states can be specified for a number of nuclei. Each nucleus has its own subsection which starts with the specification of charge Z and mass number A, and ends with the label NUCEND (or END for the last nucleus in the list).
- After the Z-A identification of the nucleus, a list of nuclear states follows with one entry per line, giving excitation energy (in MeV), spin (in multiples of 1/2), and parity (positive/even parity when the spin integer is >= zero, negative/odd parity with spin < zero).
- The ground state (zero excitation energy) has to be included!
- There is an upper limit of how many states (including g.s.) can be specified. The value depends on the code version you are using. More states will not trigger an error message but will simply be ignored.
- If no states are explicitely specified for a given nucleus, the default states (from Table of Isotopes) are used. Otherwise, the default levels are superseded by the ones in the input file.
- Breit-Wigner Resonances:
NON-SMOKERweb can also
calculate Breit-Wigner resonance parameters and cross sections.
- For the compound nucleus of the reaction, put in excited states above the separation energy of the projectile (this only works for neutron- and proton-induced reactions). This will prompt NON-SMOKERweb to also compute and output the Breit-Wigner data.
- Most likely the other output (cross sections, rates, ...) is meaningless in this case. Remember that there still is a limit on the number of states including the g.s (see above). Try to put in as many low-lying resonances as possible, though.
- Target range:
- Section label:
TARGET_RANGE - There are two ways to specify more than one target. These are distinguished by the labels EXPLICIT and GRID, respectively, which have to appear before any data in this section. If the label EXPLICIT is found, a list of target nuclei is expected to follow, one per line. With the option GRID, a range can be defined.
- Option EXPLICIT: Specify charge Z and mass number A of a target nucleus. There can be several lines like that, each specifying another target nucleus.
- Option GRID: Specification of a Z-N range of target nuclei. The order of supplied values is Z_start N_start Z_end N_end. There can only be one line of this kind.
- Options EXPLICIT and GRID cannot be mixed.
- Section label:
- Optical potentials:
- Some hints:
- Before submitting an input file, check its validity with the checker tool! This is a C program which accepts a file name on the command line and returns more detailed error messages than the web interface (which will only tell you that there was a problem with the file). You need a C-compiler to make an executable for your architecture. Precompiled executables are provided for Linux and DEC Alpha.
- It is advisable to keep a log on what you have changed in the input file during different runs. The result page returned by NON-SMOKERweb only shows the selected options but not the content of the file, unless the "Echo file content" checkbox is selected. Even in the latter case, the whole input file is echoed, not just the selected sections.
- Since only those sections are used which have been selected in the options, sections in the input file can be selectively switched on and off without the need to remove or modify them.
- On the other hand, if you want to make changes in the input file but not in the selected options, you can just hit the reload/update button on your browser to start a new calculation.
- Access control restrictions apply equally to the options on the web page and to the input file. It is not possible to select sections in an input file for which the user does not have access permission.
- For expert users, there are also advanced options available. Do not bother with those unless you really know what you are doing!
Custom potential:
A speciality in this interface is the possibility to put in new functions for the optical potentials. If you are not satisfied with the provided options, you can provide a potential in the custom area. Here are some guidelines what to put into the fields:- Functions can be written as FORTRAN style expressions, including brackets. The input is not case sensitive.
- Whitespaces (blanks, tabs, newlines) are ignored in the evaluation of the expression. This can be used to more nicely structure the input. Note that the allowed line length in the input file may be shorter than the maximum length of the expression! For a longer expression it is strongly suggested to distribute it across several lines. Otherwise an error may result.
- The standard arithmetic operators + - * / are known.
Exponentiation
can be written either as ** or as ^. Additionally, the following
functions are known:
- exp(x)...exponentiation of Euler's number, e^x
- log(x)...natural logarithm of x
- sqrt(x)...Square root of x
- For comparison of two values, the test operator '<' can be used. (Note that there is NO '>' operator!) An expression x<y will return 1 if x is smaller than y and 0 otherwise. This can be used to switch between different potential behavior depending, e.g., on the energy. See the examples below on how to use the operator. The operator precedence is stronger than + - * / but equal to exponentiation and the functions exp, log, sqrt. It is recommended to always use brackets to ensure the right order of operator evaluation.
- Numbers can be input in various formats, e.g. 1 1. 1.0 1.e00 1.0e+00 . All numbers are treated as double precision regardless of their input format. Likewise, all computations are performed in double precision.
- If desired, an equation-like input can be used, e.g. mymath=1+sqrt(3/2). Everything to the left of the equation sign is ignored, only the RHS is used for computation.
- Four variables (not case sensitive) can be used in the
expression:
- r...potential radius in fm
- en...c.m. energy of projectile-target system in MeV (for energy-dependent potentials)
- c...Coulomb barrier in MeV
- f...Currently, this is set to be A**(1./3.). Caution: The meaning of this variable may change in the future, based on common practice.
- Examples for valid expressions:
- -40/(1+exp((r-12.5)/0.62))
- mypot=-40/(1+exp((r-12.5)/0.62))
- -4.e1/(1+exp((r-1.25*f)/0.62))
- (en<5)*(-100+5.*en)+(1-(en<5))*(-75./(1.+exp((r-12.5)/4.5)))
Applicability of the model
Please be aware that the statistical model may not be applicable below a certain energy or temperature, although values are given below that limit. The statistical model assumes that averaged transmission functions can be used, i.e. averaging over resonances in the contributing energy window is permitted. At lower excitation energy of the compound nucleus, fewer resonances will be available and thus there may be a lower energy/temperature limit for the application of the model. An estimate of the limiting energy/temperature was derived in a previous paper and is also explicitly given in ADNDT 75 (2000) 1.The applicability limits can also be checked by calling a result from the standard result database and looking for possible applicability warning messages in the output. (See also the explanations of the database.) It has to be emphasized that very conservative assumptions are used and that the given results may be valid even below the given limit. The warning is merely issued to point out that one should be cautious and aware of the limitations.
There may be another limitation inherent to all statistical
model
calculations which do not explicitly include gamma cascades in the
compound nucleus. Treating the cascades and the particle escape from
the
cascade only approximately may lead to larger errors for reactions with
negative reaction Q values, i.e. for endothermic
reactions. This
is not necessarily the case but, in general, for these cases it is
safer
to calculate the reverse reaction (with positive Q
value) and
compute the desired reaction by applying detailed balance.
Referencing the results
When using NON-SMOKERweb and the
linked pages, credit
should be given to the access
page, the
NON-SMOKER papers,
and the author.
Do not forget to quote the version number you used because
different versions may yield different results. The version
number is given at the top of each result page returned by the web interface.
The desired way of quoting would be, e.g., "...obtained with NON-SMOKER(WEB) v5.9.1w...".
I would appreciate it if a copy of the work is
sent to me (e.g. at  )
before
publication, so that misconceptions or misinterpretations of the
results
can be avoided.
)
before
publication, so that misconceptions or misinterpretations of the
results
can be avoided.
Please see also the disclaimer.
Error messages
There are several error messages which can be returned
instead of the
requested result. They should be self-explanatory. In addition to
errors arising from access control or incorrect specifications in the
target options, there can be fatal errors when using the customized
potential
option. These errors will be shown at the end of an incomplete result
page
and they always refer to a problem with the functions entered in the
custom
area. Please check your input carefully!
Bug reports
If you encounter a failure or an inexplicable result, please
first check
the explanation of NON-SMOKERweb
and the used methods.
Consult the history section for
further details
of the current release.
In case you still think that there might be a bug in the returned result, please send a bug report by email, specifying exactly what you did and where you see the problem.
Thomas Rauscher
2. History
24-Aug-2006
This history list is being discontinued. Look in the forum for information on updates and revisions.20-Aug-2006
Migration to new server and layout change in the web pages. The function of the code should be unaffected.01-Jun-2006 to 02-Jun-2006
Server temporarily unavailable due to scheduled power shutdown.01-Apr-2006
Version 3.3w: Minor changes and bug fixes.09-Jul-2005 to 11-Jul-2005
Temporarily unavailable due to hardware problem!Fixed.
15-Mar-2005
- Version 3.2w: Add Coulomb radius parameter in definition of external potential.
- Requires new sample input files.
- New version of checker (Websmokertools Version 2.2) to accommodate the above change.
- Additional advanced option (deformation).
1-Mar-2005
Version 3.1w: New advanced option and bug fix (specification of external potential).25-Feb-2005
Version 3.0w: File upload from any directory readable by the user (external file does not have to be in a WWW directory anymore). Some minor bug fixes.24-Feb-2005
Switched to HTTP POST for sending the form. That means that the result page cannot be bookmarked or linked anymore!21-Feb-2005
Version 2.01w. Some bug fixes concerning inclusion of experimental masses and custom potentials.25-Oct-2004
Version 2.0w. Update of experimental masses. Now the experimental and recommended masses from the Audi mass evaluation 2003 are used.7-Oct-2004
Version 1.1w. New options for custom input file.22-Sep-2004
Going on-line with version 1.0w!This version also includes E2 gamma-transitions (the published standard results only use E1 and M1).